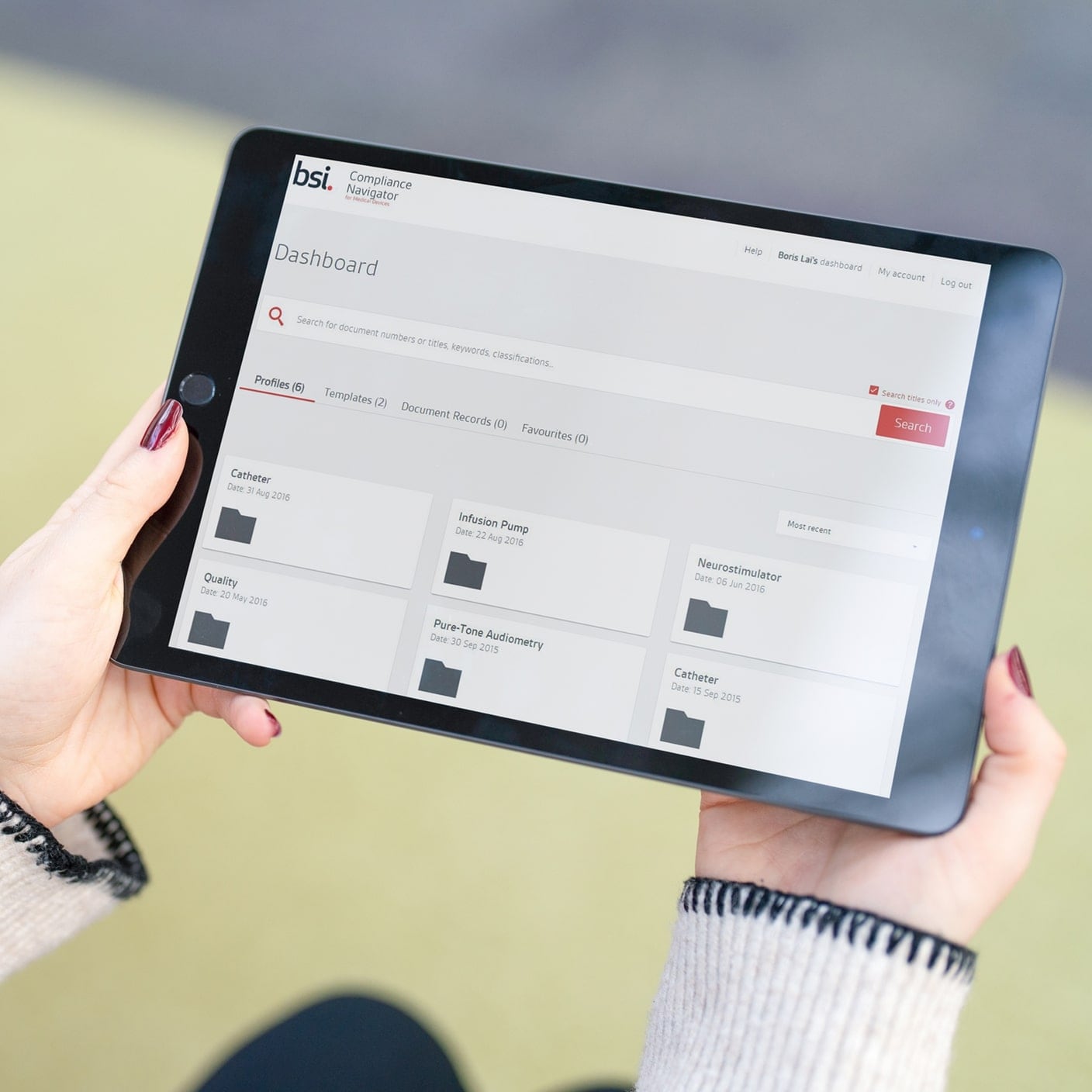
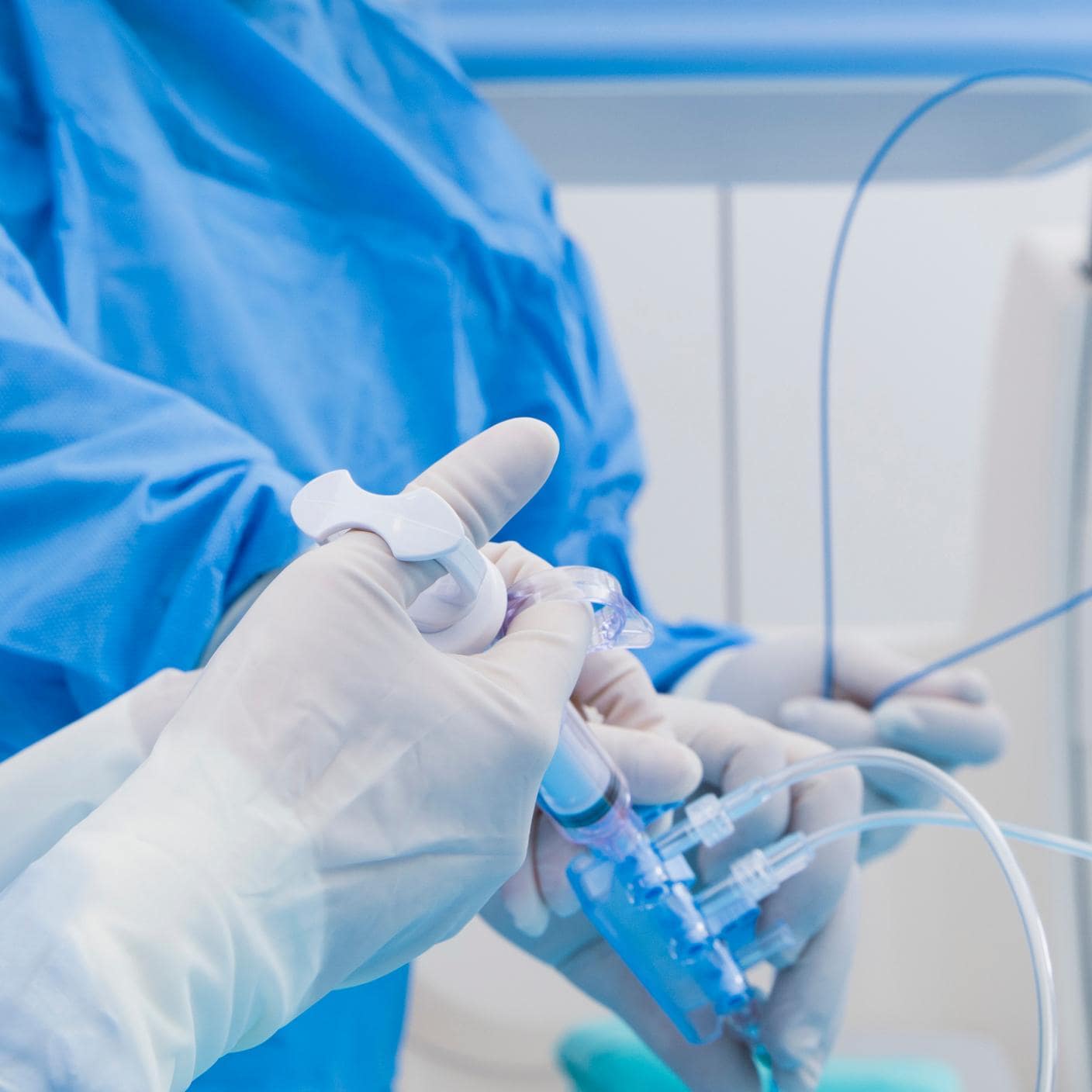
We guide you on your journey to meet global standards, promote safety, and enhance trust with your clients. As the world’s first national standards body, our unrivalled global expertise and long-standing commitment to public benefit and innovation sets the benchmark for regulatory excellence.
Back to menu
Choose a country/region
- Australia - English
- Austria - Deutsch
- Belgium - Nederlands
- Brazil - Português
- Canada - English
- Canada - Français
- Chile - Español
- China - 简体中文
- Colombia - Español
- Costa Rica - Español
- Czech Republic - English
- France - Français
- Germany - Deutsch
- Hong Kong (SAR China) - English
- India - English
- Indonesia - English
- Ireland - English
- Israel - English
- Italy - Italiano
- Japan - 日本語
- Korea - 한국어
- Malaysia - English
- MEA - عربي
- MEA - English
- Mexico - Español
- Mongolia - English
- Netherlands - English
- Netherlands - Nederlands
- New Zealand - English
- Peru - Español
- Philippines - English
- Poland - Polski
- Singapore - English
- South Africa - English
- Spain - Español
- Sweden - English
- Switzerland - Deutsch
- Taiwan - 繁體中文
- Thailand - ไทย
- Turkey - Türkçe
- United States - English
- Vietnam - English
- Vietnam - Tiếng Việt
Suggested region and language based on your location
Your current region and language