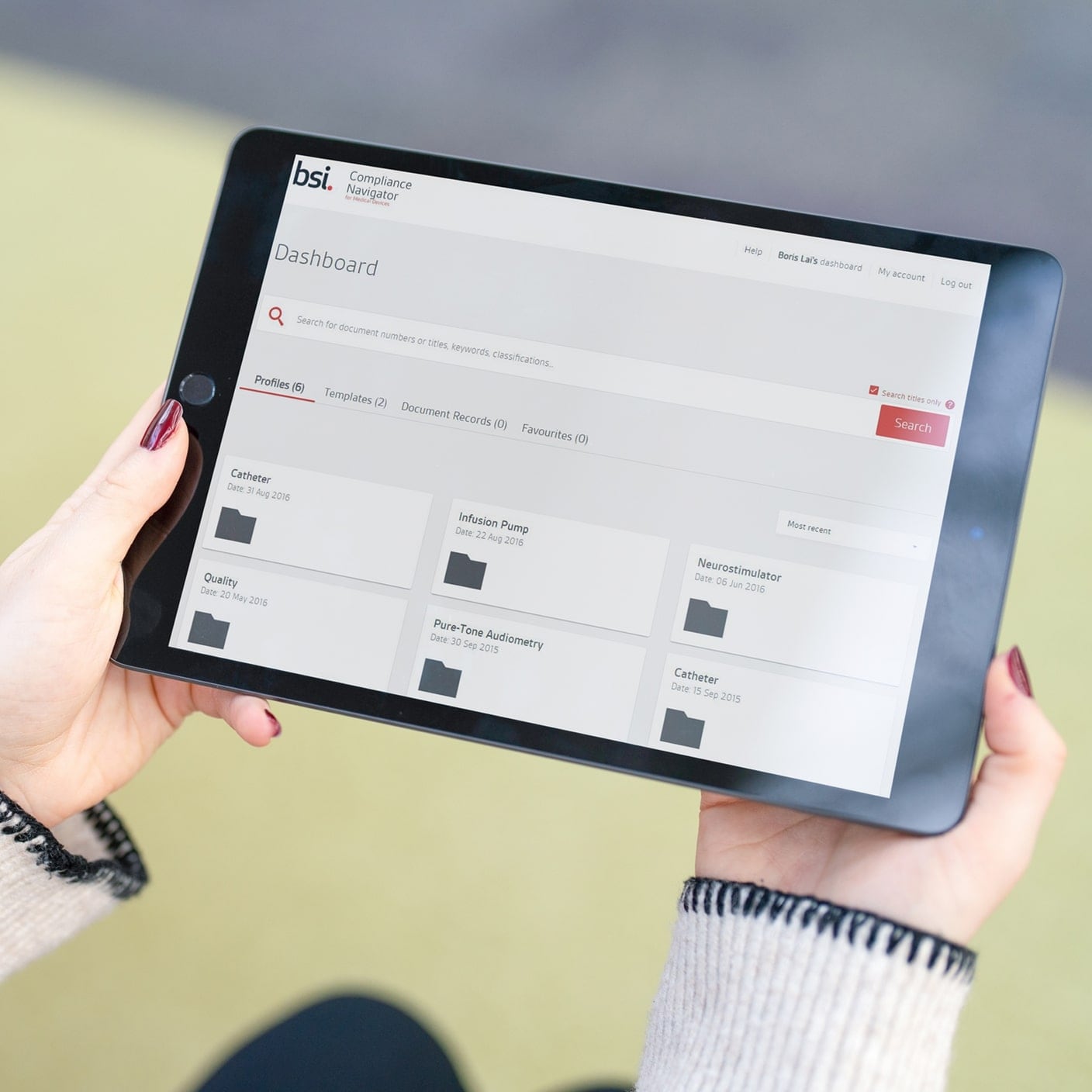
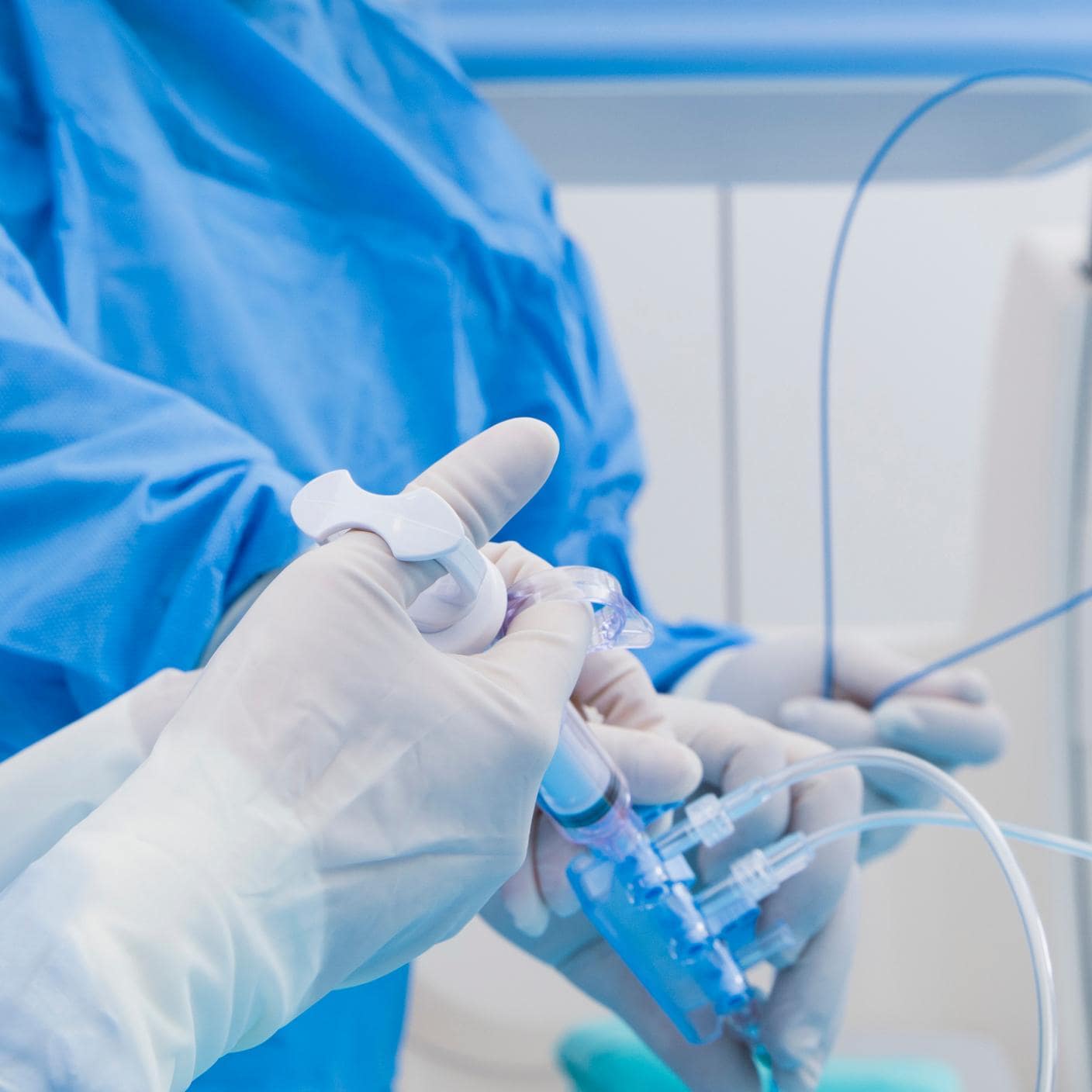
We impartially drive innovation, promote safety and enhance trust in compliance. We guide you on your journey to meet global standards, promote safety, and enhance trust with your clients. As the world’s first national standards body, our unrivalled global expertise and long-standing commitment to public benefit and innovation sets the benchmark for regulatory excellence.
Back to menu
Choose a country/region
- Australia - English
- Austria - Deutsch
- Belgium - Nederlands
- Brazil - Português
- Canada - English
- Canada - Français
- Chile - Español
- China - 简体中文
- Colombia - Español
- Costa Rica - Español
- Czech Republic - English
- France - Français
- Germany - Deutsch
- Hong Kong (SAR China) - English
- India - English
- Indonesia - English
- Ireland - English
- Israel - English
- Italy - Italiano
- Japan - 日本語
- Korea - 한국어
- Malaysia - English
- MEA - عربي
- MEA - English
- Mexico - Español
- Mongolia - English
- Netherlands - English
- Netherlands - Nederlands
- New Zealand - English
- Peru - Español
- Philippines - English
- Poland - Polski
- Singapore - English
- South Africa - English
- Spain - Español
- Sweden - English
- Switzerland - Deutsch
- Taiwan - 繁體中文
- Thailand - ไทย
- Turkey - Türkçe
- United Kingdom - English
- Vietnam - English
- Vietnam - Tiếng Việt
Suggested region and language based on your location
Your current region and language