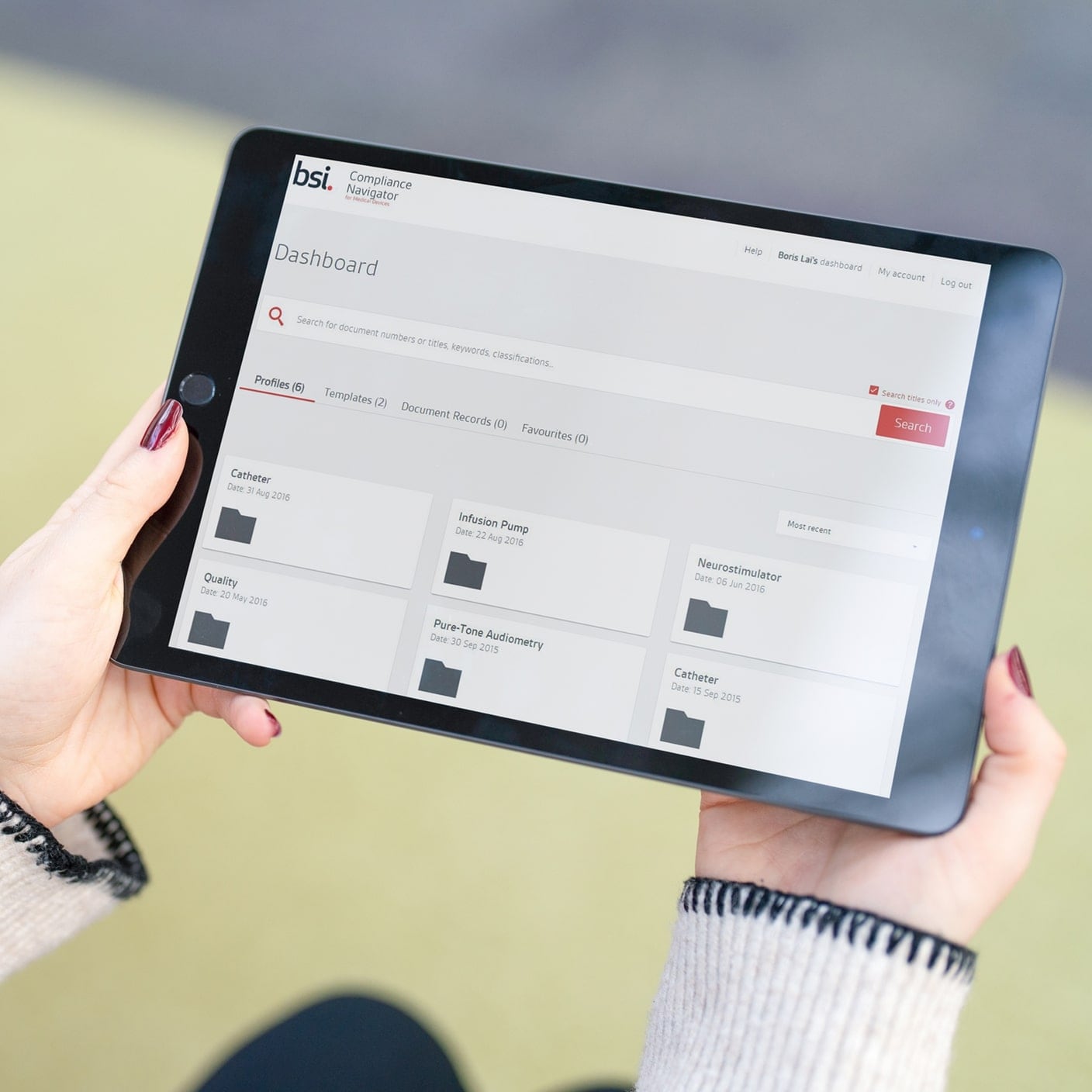
BSI hat eine Belegschaft von 5000, die von 12.000 Branchenexperten in über 193 Ländern unterstützt werden. Unsere regulatorischen Dienstleistungen und unsere weltweit führende Erfahrung bieten Ihnen effiziente Möglichkeiten, Ihr Produkt auf den Markt zu bringen.
Zurück zum Menü
Wählen Sie ein Land/eine Region
- Australia - English
- Austria - Deutsch
- Belgium - Nederlands
- Brazil - Português
- Canada - English
- Canada - Français
- Chile - Español
- China - 简体中文
- Colombia - Español
- Costa Rica - Español
- Czech Republic - English
- France - Français
- Hong Kong (SAR China) - English
- India - English
- Indonesia - English
- Ireland - English
- Israel - English
- Italy - Italiano
- Japan - 日本語
- Korea - 한국어
- Malaysia - English
- MEA - عربي
- MEA - English
- Mexico - Español
- Mongolia - English
- Netherlands - English
- Netherlands - Nederlands
- New Zealand - English
- Peru - Español
- Philippines - English
- Poland - Polski
- Singapore - English
- South Africa - English
- Spain - Español
- Sweden - English
- Switzerland - Deutsch
- Taiwan - 繁體中文
- Thailand - ไทย
- Turkey - Türkçe
- United Kingdom - English
- United States - English
- Vietnam - English
- Vietnam - Tiếng Việt
Suggested region and language based on your location
Your current region and language