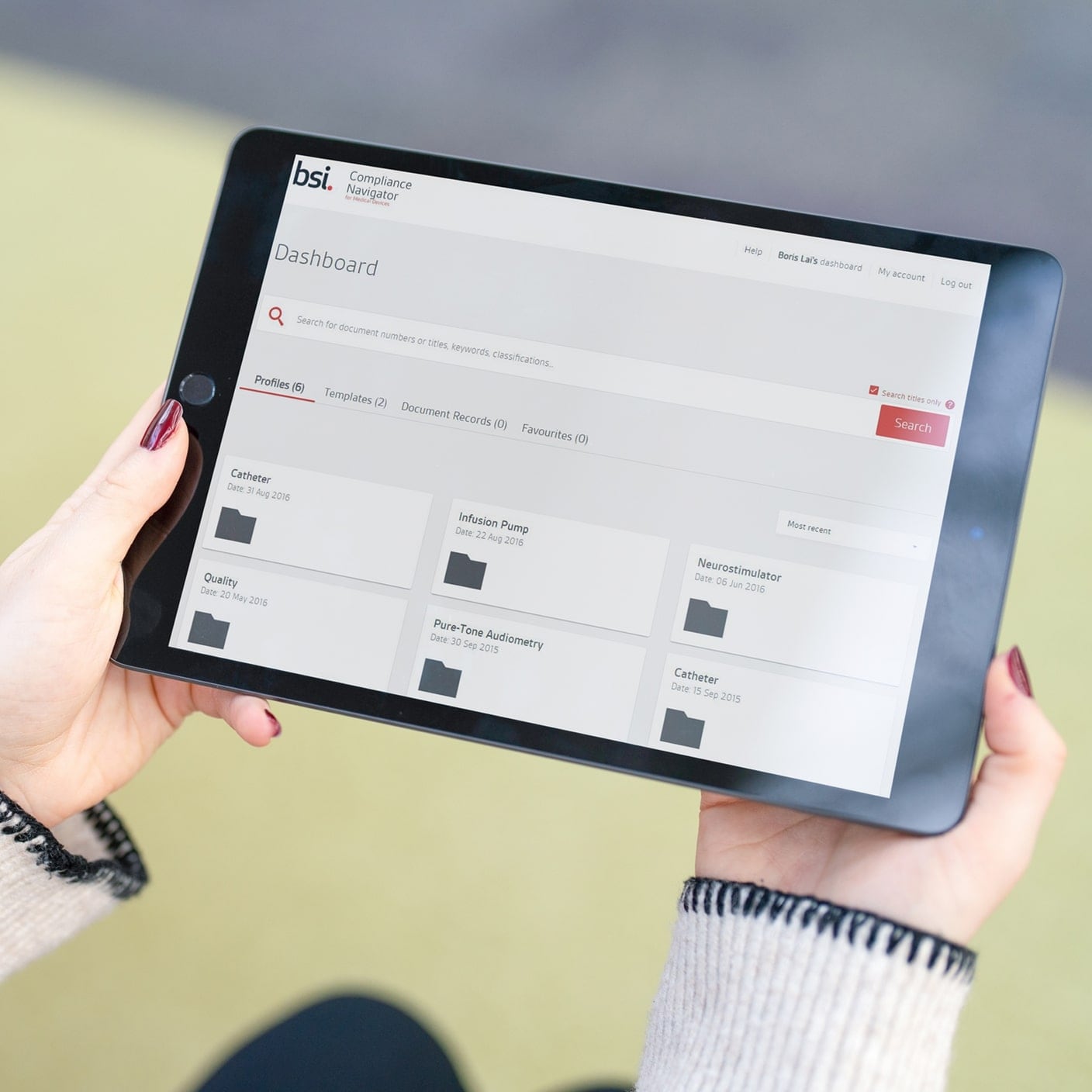
BSI está formada por 5,000 personas respaldadas por 12,000 expertos de la industria en más de 193 países. Nuestros servicios regulatorios combinados con nuestra experiencia líder mundial brindan vías eficaces para lanzar su dispositivo al mercado.
Volver al menú
Seleccionar un país o una región
- Australia - English
- Austria - Deutsch
- Belgium - Nederlands
- Brazil - Português
- Canada - English
- Canada - Français
- Chile - Español
- China - 简体中文
- Colombia - Español
- Costa Rica - Español
- Czech Republic - English
- France - Français
- Germany - Deutsch
- Hong Kong (SAR China) - English
- India - English
- Indonesia - English
- Ireland - English
- Israel - English
- Italy - Italiano
- Japan - 日本語
- Korea - 한국어
- Malaysia - English
- MEA - عربي
- MEA - English
- Mexico - Español
- Mongolia - English
- Netherlands - English
- Netherlands - Nederlands
- New Zealand - English
- Peru - Español
- Philippines - English
- Poland - Polski
- Singapore - English
- South Africa - English
- Sweden - English
- Switzerland - Deutsch
- Taiwan - 繁體中文
- Thailand - ไทย
- Turkey - Türkçe
- United Kingdom - English
- United States - English
- Vietnam - English
- Vietnam - Tiếng Việt
Suggested region and language based on your location
Your current region and language